Powered By



 Continue with Facebook
Continue with Email
Continue with Facebook
Continue with Email
Pulmonary arterial hypertension (PAH) — one type of pulmonary hypertension — can be difficult to diagnose. The disorder causes tiny blood vessels in the lungs to narrow and create high blood pressure in the lungs. Pressure in the lungs makes the right side of the heart work harder than normal. Over time, increased blood pressure in the lungs can damage the heart.
Early on, signs of PAH are unlikely to be detected during a regular physical exam, even if the disease has progressed. It may take years to diagnose PAH correctly because symptoms of PAH may be confused with other diseases.1
PAH is a rare disease, diagnosed in only 1 or 2 people in 1 million in the United States each year, according to the National Association of Rare Diseases. PAH is typically diagnosed and treated by a pulmonary hypertension specialist.1 Many types of doctors, including pulmonologists, cardiologists, and rheumatologists with specialized training in the condition, may be involved in managing PAH.2
Because of the complexity of treatment, people with PAH should be followed regularly by health care specialists. Family physicians should be made aware of any PAH treatments prescribed to avoid drug interactions and monitor potential adverse effects.3
Read more about symptoms, causes, and risk factors for PAH in What Is Pulmonary Arterial Hypertension?
 | How Is Pulmonary Arterial Hypertension Diagnosed? |
Since signs and symptoms of PAH can be subtle in early stages of the condition, a high degree of suspicion is necessary to diagnose PAH.4 A wide range of tests and exams may first be used to rule out other disorders — such as asthma, emphysema, and chronic obstructive pulmonary disease (COPD) — and then identify the type and severity, or functional class, of pulmonary arterial hypertension. These tests include:
 Physical examination — A thorough physical exam and medical history will be conducted by your doctor, often in conjunction with a cardiologist and pulmonologist. They will ask about symptoms, screen for risk factors, and listen to the heart and lungs. They will also check for swelling, changes in skin color, and other symptoms of PAH. Results of the physical exam may suggest PAH, another type of pulmonary hypertension, or a separate condition.5
Physical examination — A thorough physical exam and medical history will be conducted by your doctor, often in conjunction with a cardiologist and pulmonologist. They will ask about symptoms, screen for risk factors, and listen to the heart and lungs. They will also check for swelling, changes in skin color, and other symptoms of PAH. Results of the physical exam may suggest PAH, another type of pulmonary hypertension, or a separate condition.5
 Blood tests — The blood contains substances that can indicate PAH or other health conditions. A range of blood tests can check oxygen levels and screen for other conditions that can lead to PAH, such as heart failure, HIV infection, thyroid problems, and autoimmune diseases.5 Iron studies may also be conducted. A 2011 study found a high prevalence of iron deficiency in patients with idiopathic pulmonary arterial hypertension (IPAH), or PAH with no known cause.6 While blood tests cannot diagnose PAH, they can help assess the likelihood that your symptoms are caused by PAH.
Blood tests — The blood contains substances that can indicate PAH or other health conditions. A range of blood tests can check oxygen levels and screen for other conditions that can lead to PAH, such as heart failure, HIV infection, thyroid problems, and autoimmune diseases.5 Iron studies may also be conducted. A 2011 study found a high prevalence of iron deficiency in patients with idiopathic pulmonary arterial hypertension (IPAH), or PAH with no known cause.6 While blood tests cannot diagnose PAH, they can help assess the likelihood that your symptoms are caused by PAH.
 Imaging tests — A range of imaging tests may be ordered to determine how well the heart and lungs are functioning. Results are often key for confirming PAH as a diagnosis. Some of these tests are listed in the chart below.
Imaging tests — A range of imaging tests may be ordered to determine how well the heart and lungs are functioning. Results are often key for confirming PAH as a diagnosis. Some of these tests are listed in the chart below.

 Exercise tests — A six-minute walk test can be helpful in assessing PAH since most symptoms occur with exertion.13 During a six-minute walk test, the subject walks on a treadmill while oxygen levels (and breathing) are measured. Another exam, called a cardiopulmonary exercise test (CPET or CPX), measures heart and lung function during exercise and at rest.9
Exercise tests — A six-minute walk test can be helpful in assessing PAH since most symptoms occur with exertion.13 During a six-minute walk test, the subject walks on a treadmill while oxygen levels (and breathing) are measured. Another exam, called a cardiopulmonary exercise test (CPET or CPX), measures heart and lung function during exercise and at rest.9
 Pulmonary function test (PFT) — A PFT measures how much air the lungs can hold, as well as airflow in and out of the lungs. Taking a PFT involves breathing into a tube called a spirometer. A PFT also allows doctors to check for lung diseases such as asthma, chronic obstructive pulmonary disease (COPD), or pulmonary fibrosis, which can cause blood vessel narrowing.9
Pulmonary function test (PFT) — A PFT measures how much air the lungs can hold, as well as airflow in and out of the lungs. Taking a PFT involves breathing into a tube called a spirometer. A PFT also allows doctors to check for lung diseases such as asthma, chronic obstructive pulmonary disease (COPD), or pulmonary fibrosis, which can cause blood vessel narrowing.9
 Genetic tests — If you have a family history of pulmonary hypertension or PAH, your doctor might screen for genes linked to those conditions. Doctors may recommend genetic counseling if more than two family members have PAH.14
Genetic tests — If you have a family history of pulmonary hypertension or PAH, your doctor might screen for genes linked to those conditions. Doctors may recommend genetic counseling if more than two family members have PAH.14
 | How Is PAH Treated? |
Pulmonary arterial hypertension treatment is designed to relieve symptoms and slow progression of the disease. Without treatment, PAH can lead to dangerous and potentially fatal health complications including heart failure or sudden cardiac death.6
Treatment for PAH is very individualized and depends on the type of PAH, severity of symptoms (functional class), test findings, age, other health conditions and medications taken, and home support systems. A PAH treatment regimen typically includes medication, oxygen to help breathing, pulmonary rehabilitation, and in more severe cases, lung transplantation.9
PAH treatments are prescribed to open up blood vessels in the lungs, improve blood flow from lungs to heart and body, and ultimately reduce strain on the heart. Medications may be taken by mouth, inhaled, or administered via continuous pump infusion.8 Medications are also prescribed in a variety of combinations.12
Several medications approved by the US Food and Drug Administration (FDA) for PAH treatment fall into four categories:
 Prostaglandins — People with PAH tend to have lower levels of prostaglandins, naturally occurring substances like hormones which prevent blood clots from forming in the body. When administered as medications, prostaglandins act as vasodilators, helping widen pulmonary arteries to allow more blood to flow through narrow lung vessels.5,15 Prostaglandins may also slow scarring of blood vessels, delaying progress of the disease.15 Prostaglandin drugs, which are typically administered via intravenous (IV) infusion, include: epoprostenol (sold as Flolan and Veletri), treprostinil (sold as Orenitram, Remodulin, and Tyvaso), and Ventavis (iloprost). Uptravi (selexipag), a prostacyclin receptor agonist, is believed to work by mimicking the effect of prostaglandins.16
Prostaglandins — People with PAH tend to have lower levels of prostaglandins, naturally occurring substances like hormones which prevent blood clots from forming in the body. When administered as medications, prostaglandins act as vasodilators, helping widen pulmonary arteries to allow more blood to flow through narrow lung vessels.5,15 Prostaglandins may also slow scarring of blood vessels, delaying progress of the disease.15 Prostaglandin drugs, which are typically administered via intravenous (IV) infusion, include: epoprostenol (sold as Flolan and Veletri), treprostinil (sold as Orenitram, Remodulin, and Tyvaso), and Ventavis (iloprost). Uptravi (selexipag), a prostacyclin receptor agonist, is believed to work by mimicking the effect of prostaglandins.16
 Endothelin receptor antagonists (ERAs) — ERAs are believed to block the activity of endothelin, a substance made by the body that causes blood vessels to constrict. Letairis (ambrisentan) and Tracleer (bosentan) may allow people with PAH to exert themselves physically without shortness of breath and walk farther on a six-minute exercise test.1,17 Opsumit (macitentan) may slow disease progression and potentially reverse some heart and lung damage caused by PAH.1
Endothelin receptor antagonists (ERAs) — ERAs are believed to block the activity of endothelin, a substance made by the body that causes blood vessels to constrict. Letairis (ambrisentan) and Tracleer (bosentan) may allow people with PAH to exert themselves physically without shortness of breath and walk farther on a six-minute exercise test.1,17 Opsumit (macitentan) may slow disease progression and potentially reverse some heart and lung damage caused by PAH.1
 Phosphodiesterase-5 (PDE-5) inhibitors — PDE-5 inhibitors are believed to lower blood pressure by relaxing pulmonary arteries and increasing blood flow to the lungs.15 In clinical studies, sildenafil (sold as Revatio and Viagra) increased the distance people with PAH walked and decreased pressure in the pulmonary artery. Adcirca (tadalafil) has also been shown to improve the ability to exercise.1
Phosphodiesterase-5 (PDE-5) inhibitors — PDE-5 inhibitors are believed to lower blood pressure by relaxing pulmonary arteries and increasing blood flow to the lungs.15 In clinical studies, sildenafil (sold as Revatio and Viagra) increased the distance people with PAH walked and decreased pressure in the pulmonary artery. Adcirca (tadalafil) has also been shown to improve the ability to exercise.1
 Soluble guanylate cyclase stimulators (SGCS) — SGCS are targeted therapies that cause blood vessels to dilate (become wider), reduce lung pressure, and improve exercise capacity in people with PAH.18 Adempas (riociguat) is the only approved SGCS for pulmonary arterial hypertension. Adempas may interact with other drugs that treat PAH, including sildenafil and tadalafil.15
Soluble guanylate cyclase stimulators (SGCS) — SGCS are targeted therapies that cause blood vessels to dilate (become wider), reduce lung pressure, and improve exercise capacity in people with PAH.18 Adempas (riociguat) is the only approved SGCS for pulmonary arterial hypertension. Adempas may interact with other drugs that treat PAH, including sildenafil and tadalafil.15
Most people with PAH will need supportive medications and treatments to improve function and help prevent complications.
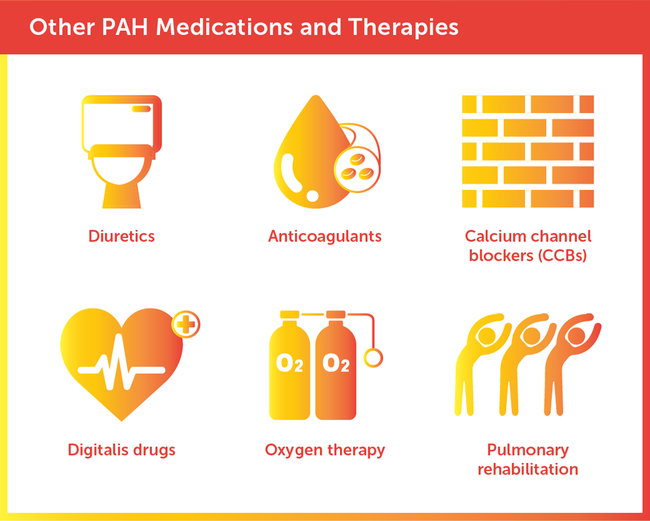
All medications have side effects, some of which are more serious than others. PAH medications may carry the risk for side effects like low blood pressure, heart complications, headache, muscle pain, indigestion, nausea, and other gastrointestinal problems.21 Treatment guidelines generally specify that safer medications should be tried first. If first-line treatments are not effective, doctors may then consider drugs with more serious side effects or higher risk for complications.22
Surgery is infrequently used as a treatment option for PAH. However, if you have severe PAH — or symptoms that are not responding to medications — your doctor may recommend one of two types of surgeries:
 Atrial septostomy — People with advanced PAH and severe right-heart failure may be candidates for atrial septostomy surgery, which can reduce symptoms. The surgeon creates a small hole between the right and left heart chambers to lower pressure on the right side. The procedure has not been clinically proven to improve survival and is rarely performed.15
Atrial septostomy — People with advanced PAH and severe right-heart failure may be candidates for atrial septostomy surgery, which can reduce symptoms. The surgeon creates a small hole between the right and left heart chambers to lower pressure on the right side. The procedure has not been clinically proven to improve survival and is rarely performed.15
![]() Transplant surgery — Lung transplant or heart-lung transplant may be options for some people with progressed PAH, or those for whom medical therapy is no longer effective. Transplant can involve replacing one or both diseased lungs, or heart and lungs, with organs from a donor. Transplantation is a high-risk operation that can result in severe complications such as organ rejection or infection.15 Transplant also can involve long waits for donor organs. In 2019, only 2,714 lung transplants and 45 heart-lung transplants were performed in the United States.23
Transplant surgery — Lung transplant or heart-lung transplant may be options for some people with progressed PAH, or those for whom medical therapy is no longer effective. Transplant can involve replacing one or both diseased lungs, or heart and lungs, with organs from a donor. Transplantation is a high-risk operation that can result in severe complications such as organ rejection or infection.15 Transplant also can involve long waits for donor organs. In 2019, only 2,714 lung transplants and 45 heart-lung transplants were performed in the United States.23
Lung transplant is complex decision — and last resort — that should be made alongside family, caregivers, and a PAH medical and transplant team.9 People with PAH have a poorer postoperative course and higher mortality rate than other lung transplant recipients, according to a report published by the American Academy of Family Physicians. Three and five-year survival rates after transplantation in those with PH are 55 percent and 45 percent, respectively.3
Many new medications and therapies for PAH are being studied. Your doctor may suggest participating in a clinical trial to obtain access to a new treatment.8
One promising new therapy for PAH is called vagus nerve stimulation (VNS).The vagus nerve controls several body functions, including contraction and relaxation of the heart. VNS uses a device that stimulates the nerves using electrical pulses. In the past, VNS has been used to treat different types of heart disorders, including arrhythmia, heart failure, and high blood pressure.24
VNS may be useful for treating PAH because it helps normalize heart function, promotes widening of blood vessels in the lungs, suppresses inflammation, and restores the balance between the sympathetic and parasympathetic nervous system. VNS is currently being tested for PAH in animal studies.24
Get updates directly to your inbox.





 Continue with Facebook
Continue with Email
Continue with Facebook
Continue with Email
Continue with Facebook
Continue with Email
Continue with Facebook
Continue with Email
Become a member to get even more





 Join
Join


 Your Privacy Choices
Your Privacy Choices

This is a member-feature!
Sign up for free to view article comments.
This is great information to know especially with the corona virus. Great information on how to take better care of ourselves.
We'd love to hear from you! Please share your name and email to post and read comments.
You'll also get the latest articles directly to your inbox.